機械学習アルゴリズムを用いてDNAシーケンスを高速化 韓国KAIST
韓国科学技術院(KAIST)は5月10日、同院の研究チームが、機械学習手法を用いて、DNAシーケンスにおけるショートリードのアライメント(alignment)プロセスを大幅に高速化できるソフトウェアを開発したと発表した。この研究の成果は3月7日付で学術誌 Bioinformatics に報告された。
最新の次世代シーケンス(NGS)ハードウェアはDNA配列の断片(ショートリード)数十億個を1回のランで生成できる。しかし、人口規模のゲノムシーケンシングを実現するには、これらの大量のショートリードを参照DNAシーケンスと整列させるアライメントを効率化する必要がある。
ショートリードのアライメントによく用いられるソフトウェア「BWA-MEM2」では、アライメントプロセスのうち、ショートリードの完全一致を探す「seeding」が高速化のボトルネックとなっていた。
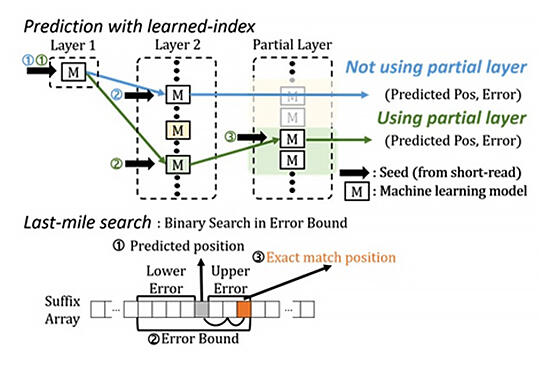
機械学習でDNAシーケンスを高速化するアプローチ (提供:KAIST)
研究チームは、学習型インデックス(learned index)と呼ばれる機械学習手法を用いてBWA-MEM2のアルゴリズムを改良し、命令数とメモリアクセスを削減してseedingを3.45倍高速化することに成功した。ハン・ドンス(Han Dongsu)教授は「機械学習を利用して、ゲノムのビッグデータの完全な解析を従来の手法より高速かつ低コストで行えることが示された」と語った。
研究チームは、学術機関や産業界の科学者がゲノミクスのビッグデータ解析に日常的に利用できる効率的なソフトウェアを開発することを目標としている。
サイエンスポータルアジアパシフィック編集部


